Más cerca de entender la enfermedad congénita del metabolismo más frecuente
Un equipo internacional con participación de científicos del Consejo Superior de Investigaciones Científicas (CSIC) ha logrado determinar la estructura atómica de la fenilalanina hidroxilasa, una enzima humana cuyos fallos son responsables del error innato del metabolismo más frecuente: la fenilcetonuria. Los resultados de este trabajo aparecen publicados en la revista Proceedings of the National Academy of Sciences (PNAS).
La fenilcetonuria, también conocida como PKU, es una enfermedad congénita que afecta a uno de cada 10.000 recién nacidos. Los niños afectados con PKU sufren un trastorno de su metabolismo, ya que el cuerpo no es capaz de metabolizar adecuadamente un aminoácido, la fenilalanina, la cual, en grandes cantidades, resulta tóxica para el sistema nervioso central. Los niños afectados experimentan graves problemas de desarrollo neurológico y psicosociales que resultan en una baja calidad de vida y una gran carga social.
“Sabíamos que esta enfermedad era el resultado de mutaciones en el gen de la fenilalanina hidroxilasa o PAH, una enzima hepática que elimina el exceso de la fenilalanina que ingerimos en la dieta. Sin embargo, hasta ahora no se había determinado su estructura, la cual nos da pistas sobre su mecanismo catalítico”, explica el investigador del CSIC Juan Antonio Hermoso, del Instituto de Química Física Rocasolano, que ha dirigido el estudio junto con científicos de la Universidad de Bergen (Noruega).
La enzima PAH requiere de un mecanismo complejo de regulación: debe ser muy activa durante las comidas, a fin de eliminar el exceso de fenilalanina, pero muy poco activa entre comidas, cuando no se necesita la eliminación del aminoácido. Para ello, la enzima PAH se une a BH4, una molécula que es esencial para que se produzca la reacción enzimática, pero que también estabiliza la estructura compleja de la enzima y disminuye su actividad cuando no se necesita la eliminación del aminoácido.
“Demasiado aminoácido causa daño cerebral y muy poco dificulta nuestra propia producción de proteínas y, por lo tanto, la molécula BH4 es una de las sofisticadas formas en las que el cuerpo regula la actividad de la PAH”, detalla Hermoso.
Primera estructura completa de la PAH humana
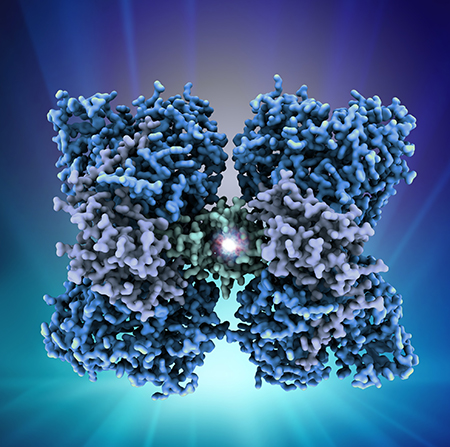
Cómo se produce esta regulación a nivel molecular es lo que la comunidad científica llevaba décadas tratando de determinar. Ahora se han descubierto las bases moleculares de este mecanismo. “Mediante cristalografía de rayos X y microscopía electrónica, hemos obtenido las estructuras tridimensionales de la enzima PAH sola y en complejo con BH4, lo que ha permitido diseccionar el mecanismo por el cual esta enzima regula su actividad en respuesta a los niveles de fenilalanina”, agrega el investigador del CSIC.
Las estructuras resultantes son las primeras estructuras completas de la enzima humana y también las primeras de una PAH completa unida al BH4. La mayoría de las mutaciones asociadas a la PKU (más de 950 según las estadísticas disponibles sobre enfermedades neurotransmisoras pediátricas) conducen a conformaciones de la PAH disfuncionales y menos estables, más propensas a degradarse.
“La propiedad estabilizadora de la molécula BH4 ha sido explotada clínicamente y es, hoy en día, la única opción de tratamiento disponible para la PKU, que ayuda a aproximadamente el 25% de los pacientes. El análisis estructural y funcional presentado ahora es importante, tanto para la comprensión del funcionamiento normal de la enzima, como para el desarrollo de nuevos tratamientos, más efectivos, contra la fenilcetonuria”, concluye Hermoso.
- Marte Innselset Flydal, Martin Alcorlo, Fredrik Gullaksen Johannessen, Siseth Martinez-Caballero, Lars Skjærven, Rafael Fernandez-Leiro, Aurora Martinez, Juan A. Hermoso. The structure of full-length human phenylalanine hydroxylase in complex with tetrahydrobiopterin. PNAS. DOI: 10.1073/pnas.1902639116.
Noticias relacionadas
Un consorcio europeo donde participan investigadores del Consejo…
Una investigación desarrollada por un equipo científico del Consejo…
Los métodos físicos de terapia del cáncer se utilizan ampliamente en la…